Abstract
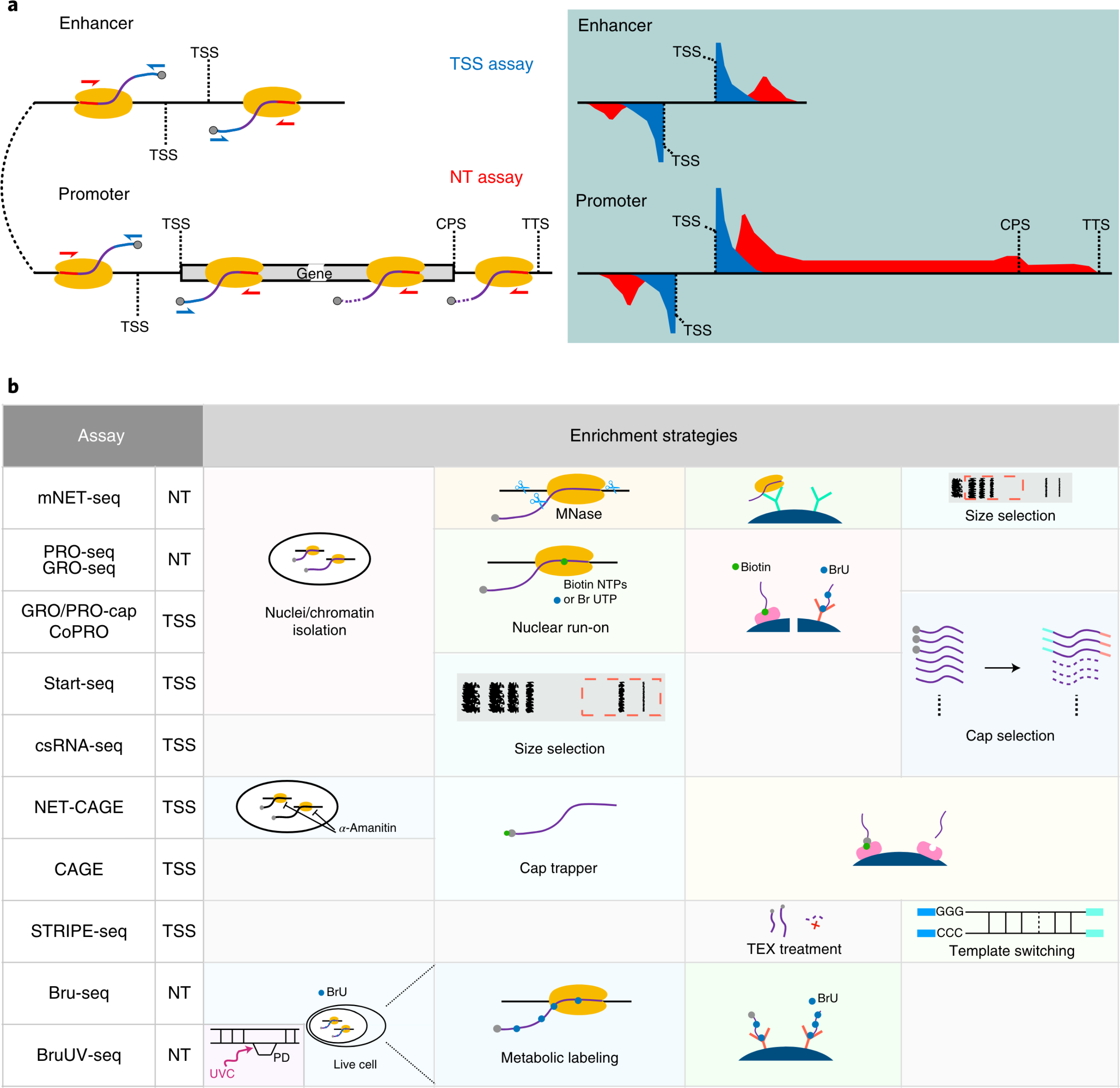
Mounting evidence supports the idea that transcriptional patterns serve as more specific identifiers of active enhancers than histone marks; however, the optimal strategy to identify active enhancers both experimentally and computationally has not been determined. Here, we compared 13 genome-wide RNA-seq assays in K562 cells and show that nuclear run-on followed by cap-selection assay (GRO/PRO-cap) has advantages in enhancer RNA detection and active enhancer identification. We also introduce a tool, PINTS, to identify active promoters and enhancers genome wide and pinpoint the precise location of 5' transcription start sites. Finally, we compiled a comprehensive enhancer candidate compendium based on the detected eRNA TSSs available in 120 cell and tissue types, which can be accessed at https://pints.yulab.org. With knowledge of the best available assays and pipelines, this large-scale annotation of candidate enhancers will pave the way for selection and characterization of their functions in a time- and labor-efficient manner.
Visual story
Key figures
Comparison of currently available assays for detection of eRNAs
Use the work
Resources
Use these links to use related materials, or continue into the full paper.
Acknowledge this work
Citation
Yao, L. et al. A comparison of experimental assays and analytical methods for genome-wide identification of active enhancers. Nat Biotechnol 40, 1056–1065 (2022).
Manuscript
Accepted manuscript
Open or download the accepted manuscript.