Abstract
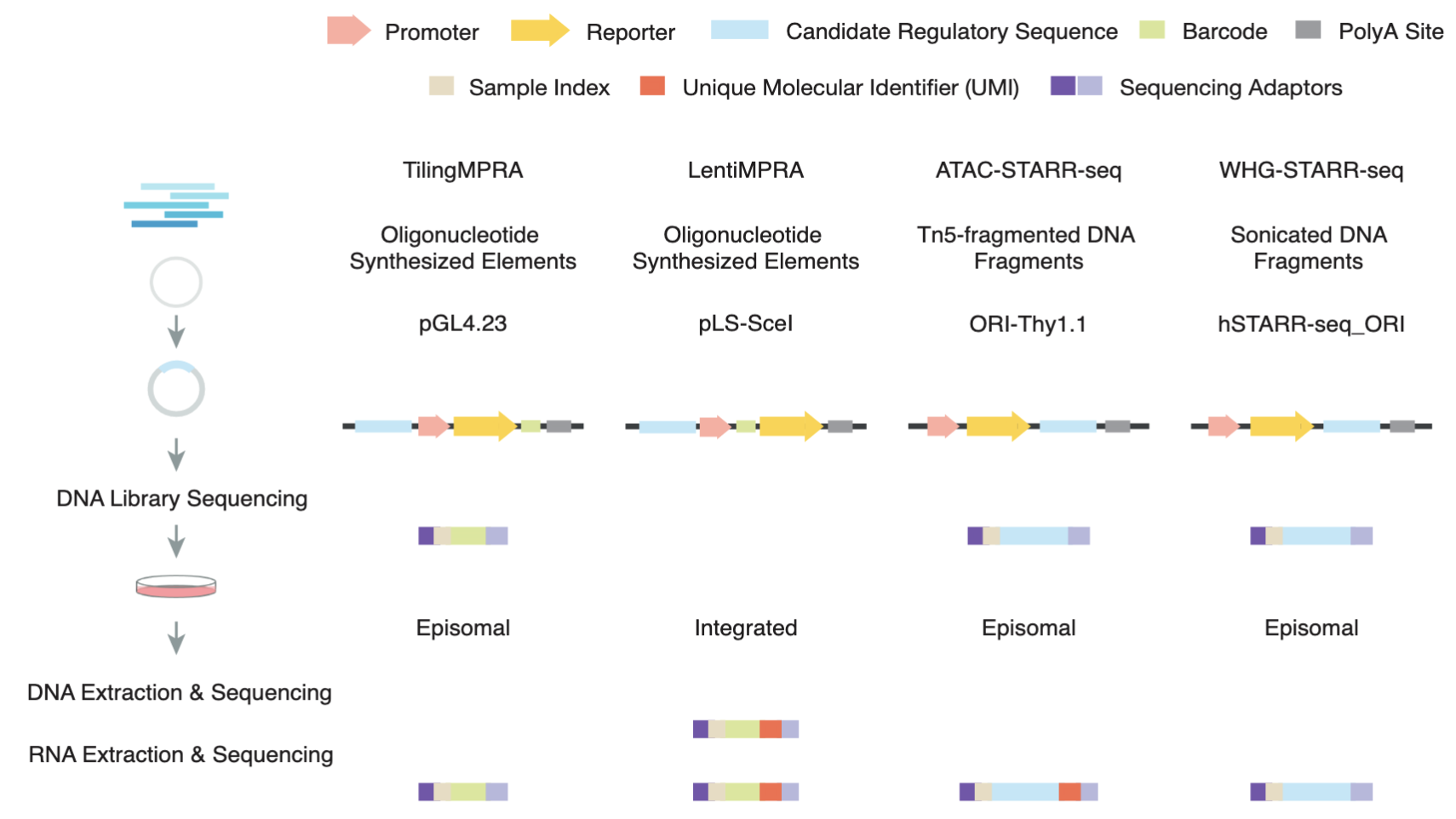
Massively parallel reporter assays (MPRAs) and self-transcribing active regulatory region sequencing (STARR-seq) have revolutionized enhancer characterization by enabling high-throughput functional assessment of regulatory sequences. Here, we systematically evaluated six MPRA and STARR-seq datasets generated in the human K562 cell line and found substantial inconsistencies in enhancer calls from different labs that are primarily due to technical variations in data processing and experimental workflows. To address these variations, we implemented a uniform enhancer call pipeline, which significantly improved cross-assay agreement. While increasing sequence overlap thresholds enhanced concordance in STARR-seq assays, cross-assay consistency in LentiMPRA was strongly influenced by assay-specific factors. Notably, our results show that LentiMPRA exhibits a strong preference for promoter-associated sequences rather than enhancers. Functional validation using candidate cis-regulatory elements (cCREs) confirmed that epigenomic features such as chromatin accessibility and histone modifications are strong predictors of enhancer activity. Importantly, our study validated transcription as a critical hallmark of active enhancers, demonstrating that highly transcribed regions exhibit significantly higher active rates across assays. Furthermore, we show that transcription enhances the predictive power of epigenomic features, enabling more accurate and refined enhancer annotation. Our study provides a comprehensive framework for integrating different enhancer datasets and underscores the importance of accounting for assay-specific biases when interpreting enhancer activity. These findings refine enhancer identification using massively parallel reporter assays and improve the functional annotation of the human genome.
Visual story
Key figures
Graphical abstract
Use the work
Resources
Use these links to use related materials, or continue into the full paper.
Acknowledge this work
Citation
Zhang J, et al. Comprehensive Evaluation of Diverse Massively Parallel Reporter Assays to Functionally Characterize Human Enhancers Genome-wide. (None). 2025; (unknown volume):(unknown pages). doi: 10.1101/2025.03.25.645321
Manuscript
Accepted manuscript
Open or download the accepted manuscript.