Abstract
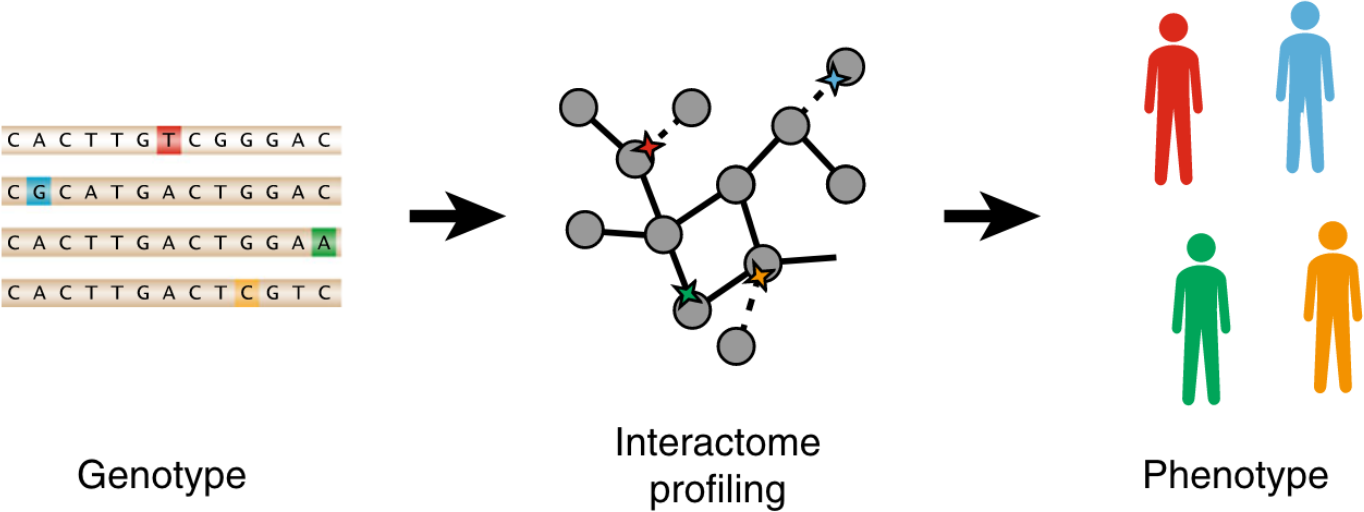
Each human genome carries tens of thousands of coding variants. The extent to which this variation is functional and the mechanisms by which they exert their influence remains largely unexplored. To address this gap, we leverage the ExAC database of 60,706 human exomes to investigate experimentally the impact of 2009 missense single nucleotide variants (SNVs) across 2185 protein-protein interactions, generating interaction profiles for 4797 SNV-interaction pairs, of which 421 SNVs segregate at > 1% allele frequency in human populations. We find that interaction-disruptive SNVs are prevalent at both rare and common allele frequencies. Furthermore, these results suggest that 10.5% of missense variants carried per individual are disruptive, a higher proportion than previously reported; this indicates that each individual's genetic makeup may be significantly more complex than expected. Finally, we demonstrate that candidate disease-associated mutations can be identified through shared interaction perturbations between variants of interest and known disease mutations.
Visual story
Key figures
Graphical abstract
Use the work
Resources
Use these links to use related materials, or continue into the full paper.
Acknowledge this work
Citation
Fragoza R, et al. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. 2019; 10:4141. doi: 10.1038/s41467-019-11959-3
Manuscript
Accepted manuscript
Open or download the accepted manuscript.