Abstract
MOTIVATION: With the rapid development of Next-Generation Sequencing, a large amount of data is now available for bioinformatics research. Meanwhile, the presence of many pipeline frameworks makes it possible to analyse these data. However, these tools concentrate mainly on their syntax and design paradigms, and dispatch jobs based on users' experience about the resources needed by the execution of a certain step in a protocol. As a result, it is difficult for these tools to maximize the potential of computing resources, and avoid errors caused by overload, such as memory overflow.
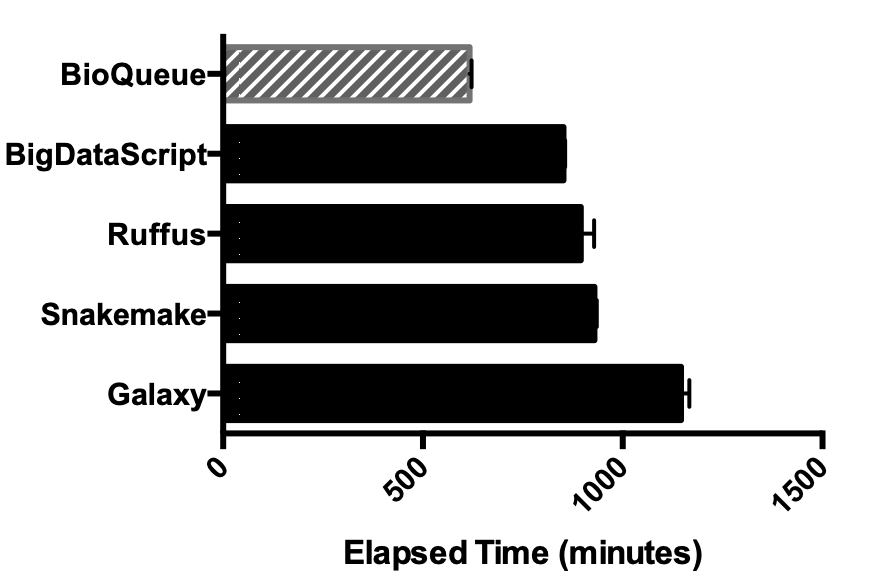
RESULTS: Here, we have developed BioQueue, a web-based framework that contains a checkpoint before each step to automatically estimate the system resources (CPU, memory and disk) needed by the step and then dispatch jobs accordingly. BioQueue possesses a shell command-like syntax instead of implementing a new script language, which means most biologists without computer programming background can access the efficient queue system with ease.
AVAILABILITY AND IMPLEMENTATION: BioQueue is freely available at https://github.com/liyao001/BioQueue. The extensive documentation can be found at http://bioqueue.readthedocs.io.
Visual story
Key figures
Graphical abstract
Pipeline execution view
A second storyline figure showing another view of the BioQueue paper assets.
Use the work
Resources
Use these links to use related materials, or continue into the full paper.
Acknowledge this work
Citation
<b>Yao, L.<sup>#</sup></b>, Wang, H., Song, Y., Sui, G.<sup>#</sup></b> BioQueue: a novel pipeline framework to accelerate bioinformatics analysis. BioQueue: a novel pipeline framework to accelerate bioinformatics analysis. 2017; 33:3286-3288. doi: 10.1093/bioinformatics/btx403
Manuscript
Accepted manuscript
Open or download the accepted manuscript.