#14: (2022). A comparison of experimental assays and analytical methods for genome-wide identification of active enhancers. Nature Biotechnology, 40(7), 1056-1065.
#13: (2019). Large-scale prediction of ADAR-mediated effective human A-to-I RNA editing. Briefings in Bioinformatics, 20(1), 102-109.
#12: (2017). BioQueue: a novel pipeline framework to accelerate bioinformatics analysis. Bioinformatics, 33(20), 3286-3288.
#11: (2023). Finding Needles in the Haystack: Strategies for Uncovering Noncoding Regulatory Variants. Annual Review of Genetics, 57(1), 201-222.
#10: (2022). Functional genomic assays to annotate enhancer-promoter interactions genome wide. Human Molecular Genetics, 31(R1), R97-R104.
#9: (2026). Dual promoter–enhancer activities reflect a unified regulatory logic. Nature Communications, 17(1), 2177.
#7: (2025). Loss of Kmt2c or Kmt2d primes urothelium for tumorigenesis and redistributes KMT2A–menin to bivalent promoters. Nature Genetics, 57(1), 165-179.
#5: (2022). Survey of the binding preferences of RNA-binding proteins to RNA editing events. Genome Biology, 23(1), 169.
#4: (2019). Inhibition of DCLK1 down-regulates PD-L1 expression through Hippo pathway in human pancreatic cancer. Life Sciences, 241 117150.
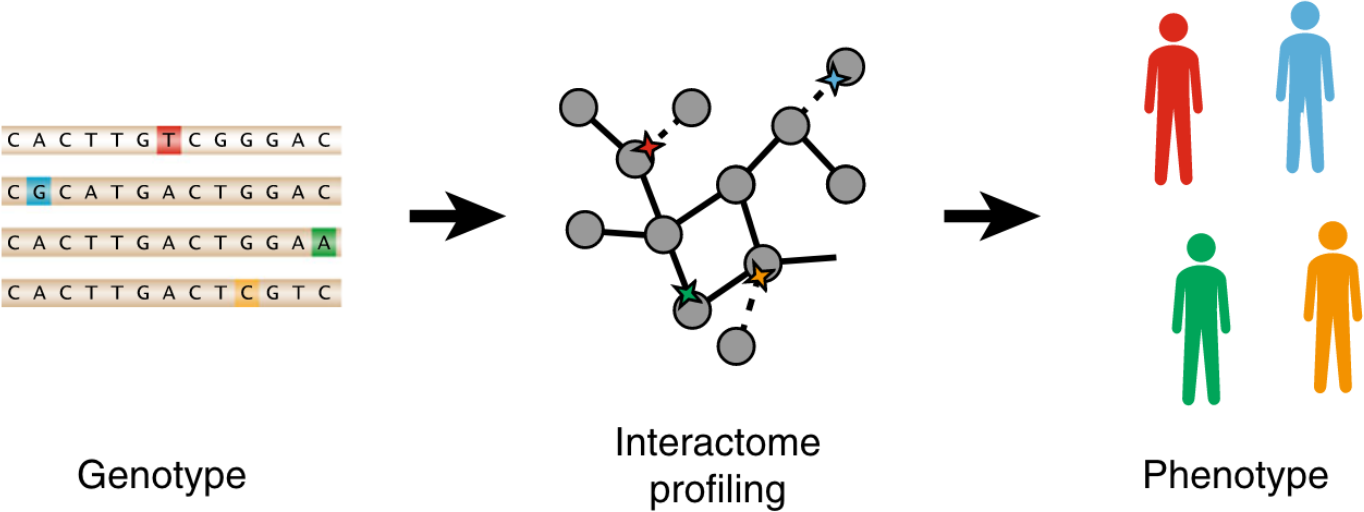
#3: (2019). Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nature Communications, 10(1), 4141.
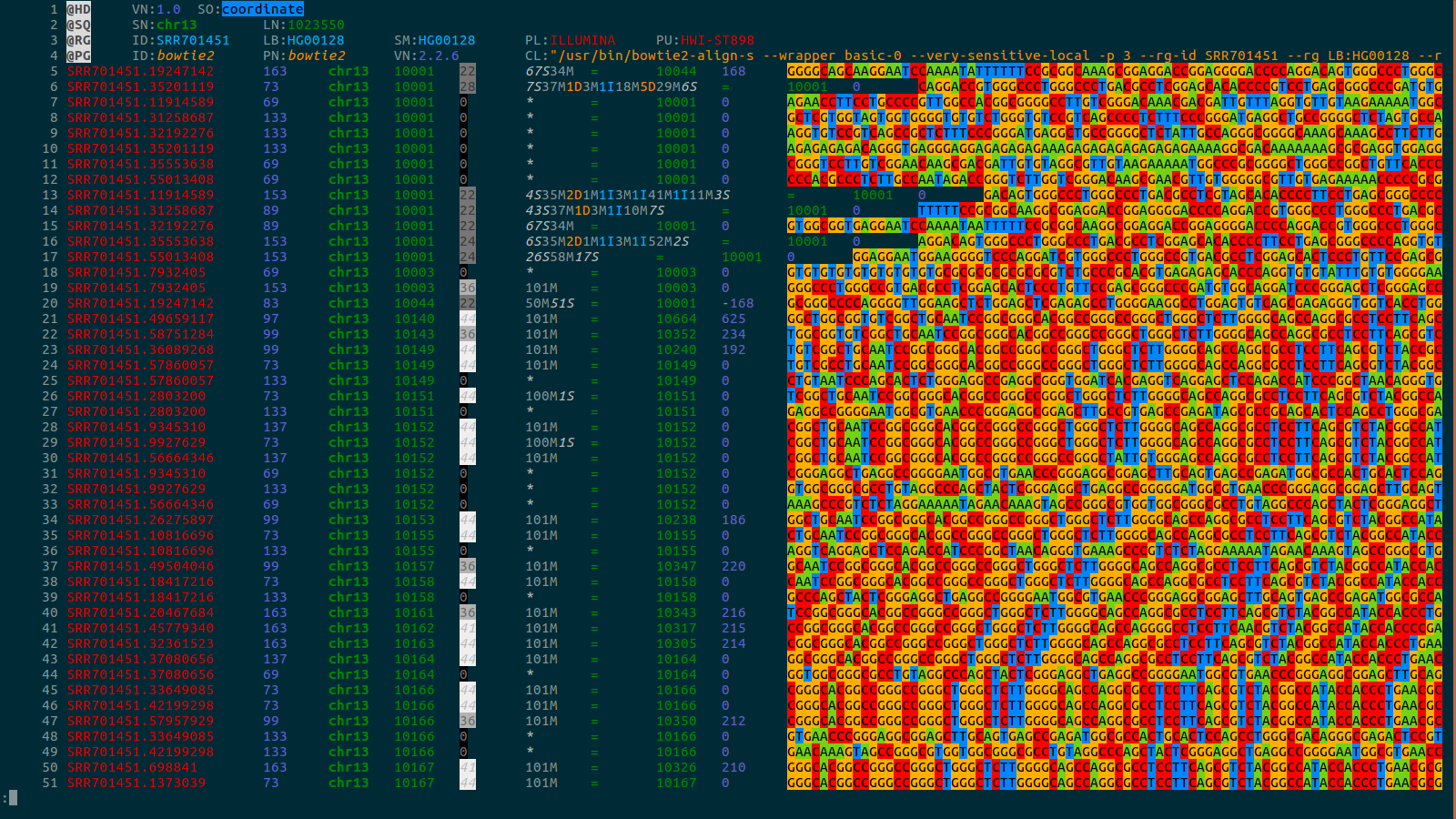
#2: (2018). bioSyntax: syntax highlighting for computational biology. BMC Bioinformatics, 19(1), 303.
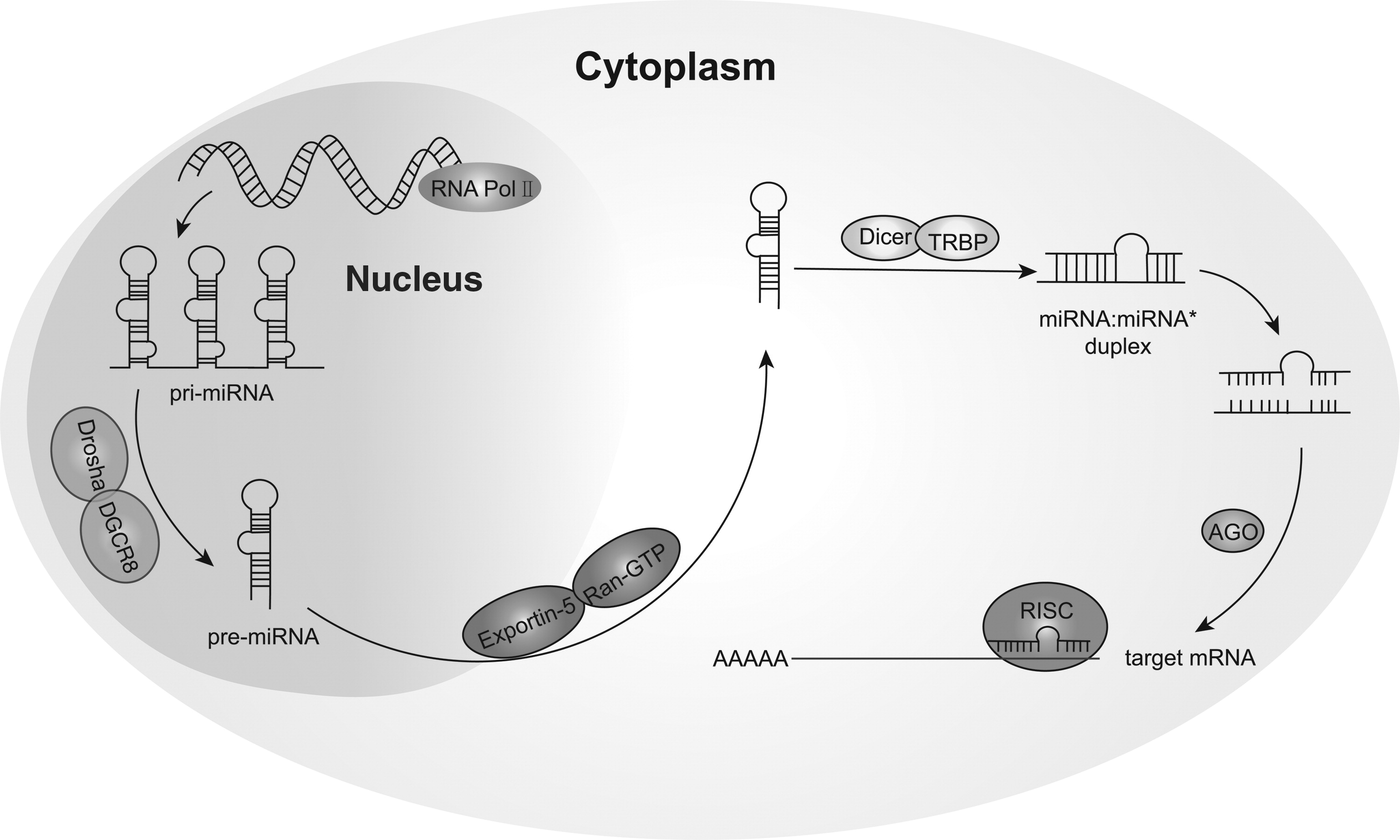
#1: (2016). Circulating microRNAs: Promising Biomarkers Involved in Several Cancers and Other Diseases. DNA and Cell Biology, 36(2), 77-94.
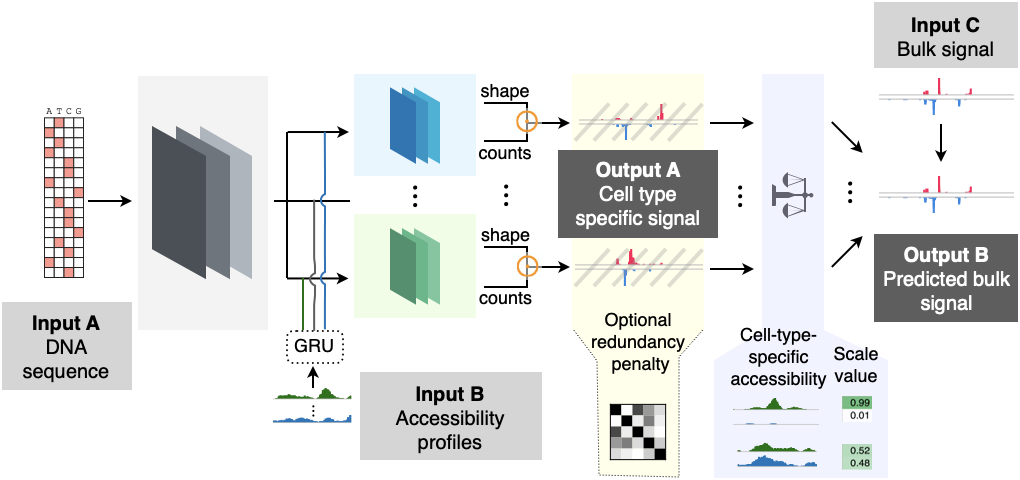
High-resolution reconstruction of cell-type specific transcriptional regulatory processes from bulk sequencing samples
Biological systems exhibit remarkable heterogeneity, characterized by intricate interplay among diverse cell types. Resolving the regulatory processes of specific cell types is crucial for delineating developmental mechanisms and disease etiologies. While single-cell sequencing methods such as scRNA-seq and scATAC-seq have revolutionized our understanding of individual cellular functions, adapting bulk genome-wide assays to achieve single-cell resolution of other genomic features remains a significant technical challenge. Here, we introduce Deep-learning-based DEconvolution of Tissue profiles with Accurate Interpretation of Locus-specific Signals (DeepDETAILS), a novel quasi-supervised framework to reconstruct cell-type-specific genomic signals with base-pair precision. DeepDETAILS’ core innovation lies in its ability to perform cross-modality deconvolution using scATAC-seq reference libraries for other bulk datasets, benefiting from the affordability and availability of scATAC-seq data. DeepDETAILS enables high-resolution mapping of genomic signals across diverse cell types, with great versatility for various omics datasets, including nascent transcript sequencing (such as PRO-cap and PRO-seq) and ChIP-seq for chromatin modifications. Our results demonstrate that DeepDETAILS significantly outperformed traditional statistical deconvolution methods. Using DeepDETAILS, we developed a comprehensive compendium of high-resolution nascent transcription and histone modification signals across 39 diverse human tissues and 86 distinct cell types. Furthermore, we applied our compendium to fine-map risk variants associated with Primary Sclerosing Cholangitis (PSC), a progressive cholestatic liver disorder, and revealed a potential etiology of the disease. Our tool and compendium provide invaluable insights into cellular complexity, opening new avenues for studying biological processes in various contexts.
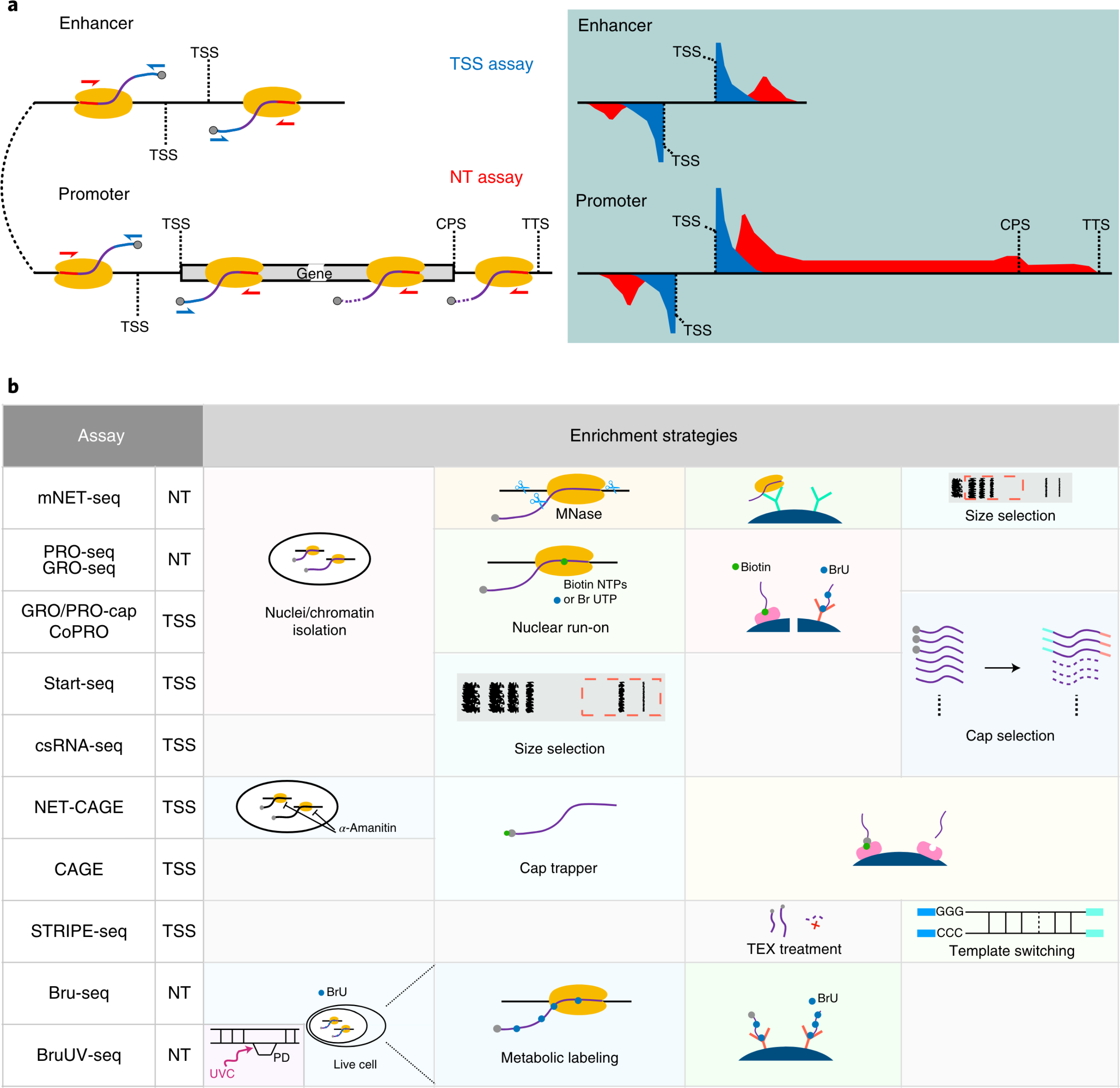
A comparison of experimental assays and analytical methods for genome-wide identification of active enhancers
Mounting evidence supports the idea that transcriptional patterns serve as more specific identifiers of active enhancers than histone marks; however, the optimal strategy to identify active enhancers both experimentally and computationally has not been determined. Here, we compared 13 genome-wide RNA-seq assays in K562 cells and show that nuclear run-on followed by cap-selection assay (GRO/PRO-cap) has advantages in enhancer RNA detection and active enhancer identification. We also introduce a tool, PINTS, to identify active promoters and enhancers genome wide and pinpoint the precise location of 5' transcription start sites. Finally, we compiled a comprehensive enhancer candidate compendium based on the detected eRNA TSSs available in 120 cell and tissue types, which can be accessed at https://pints.yulab.org. With knowledge of the best available assays and pipelines, this large-scale annotation of candidate enhancers will pave the way for selection and characterization of their functions in a time- and labor-efficient manner.
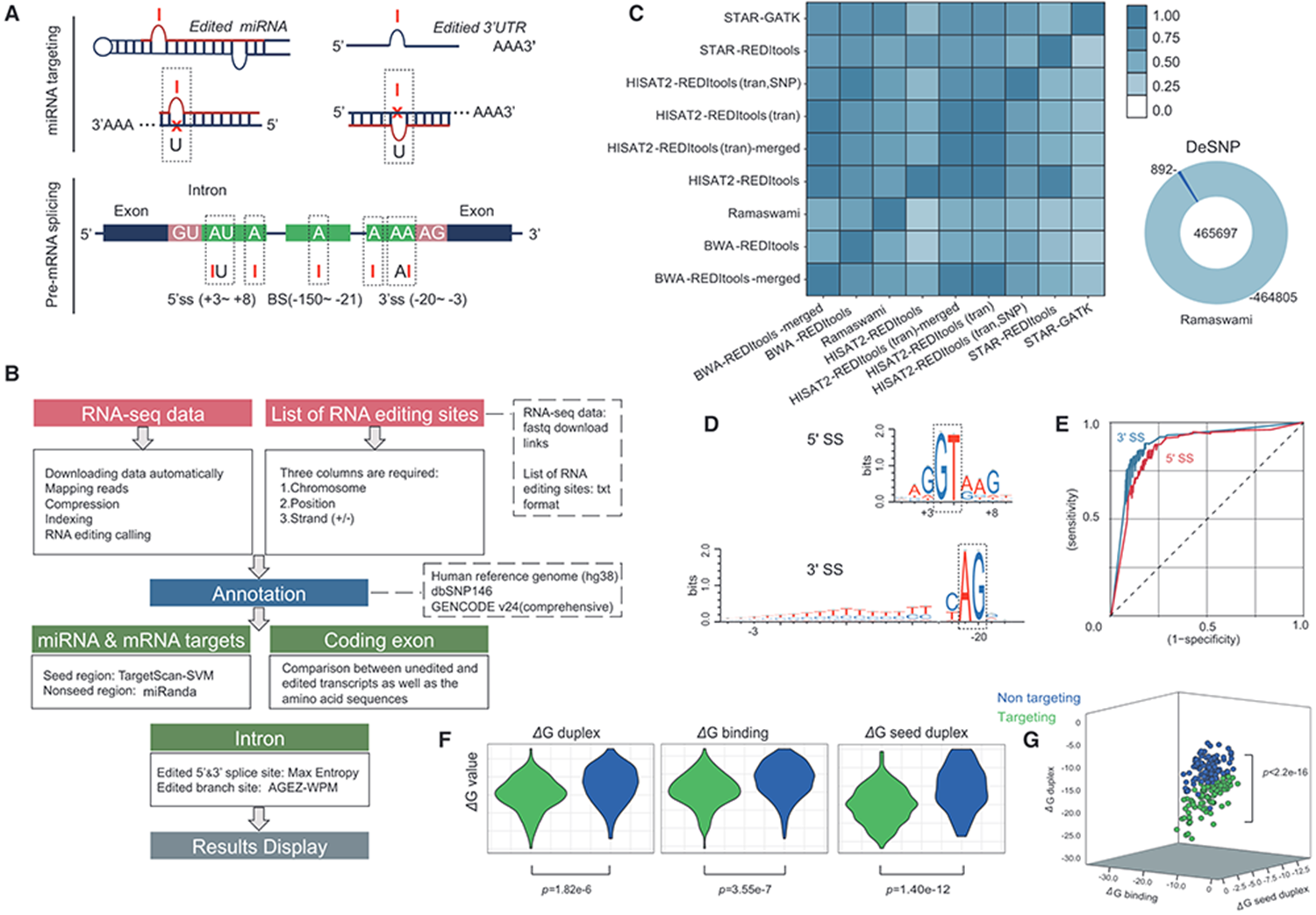
Large-scale prediction of ADAR-mediated effective human A-to-I RNA editing
Adenosine-to-inosine (A-to-I) editing by adenosine deaminase acting on the RNA (ADAR) proteins is one of the most frequent modifications during post- and co-transcription. To facilitate the assignment of biological functions to specific editing sites, we designed an automatic online platform to annotate A-to-I RNA editing sites in pre-mRNA splicing signals, microRNAs (miRNAs) and miRNA target untranslated regions (3' UTRs) from human (Homo sapiens) high-throughput sequencing data and predict their effects based on large-scale bioinformatic analysis. After analysing plenty of previously reported RNA editing events and human normal tissues RNA high-seq data, >60 000 potentially effective RNA editing events on functional genes were found. The RNA Editing Plus platform is available for free at https://www.rnaeditplus.org/, and we believe our platform governing multiple optimized methods will improve further studies of A-to-I-induced editing post-transcriptional regulation.
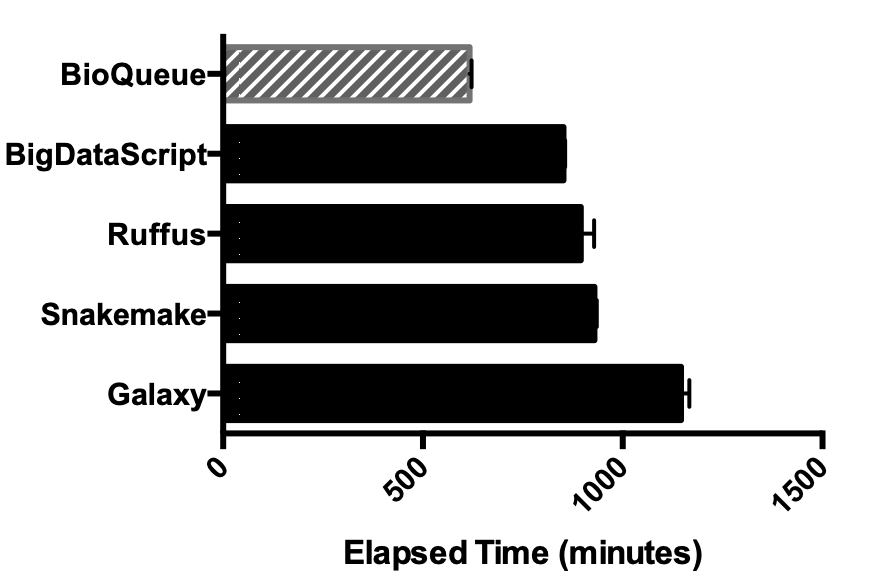
BioQueue: a novel pipeline framework to accelerate bioinformatics analysis
MOTIVATION: With the rapid development of Next-Generation Sequencing, a large amount of data is now available for bioinformatics research. Meanwhile, the presence of many pipeline frameworks makes it possible to analyse these data. However, these tools concentrate mainly on their syntax and design paradigms, and dispatch jobs based on users' experience about the resources needed by the execution of a certain step in a protocol. As a result, it is difficult for these tools to maximize the potential of computing resources, and avoid errors caused by overload, such as memory overflow.
RESULTS: Here, we have developed BioQueue, a web-based framework that contains a checkpoint before each step to automatically estimate the system resources (CPU, memory and disk) needed by the step and then dispatch jobs accordingly. BioQueue possesses a shell command-like syntax instead of implementing a new script language, which means most biologists without computer programming background can access the efficient queue system with ease.
AVAILABILITY AND IMPLEMENTATION: BioQueue is freely available at https://github.com/liyao001/BioQueue. The extensive documentation can be found at http://bioqueue.readthedocs.io.
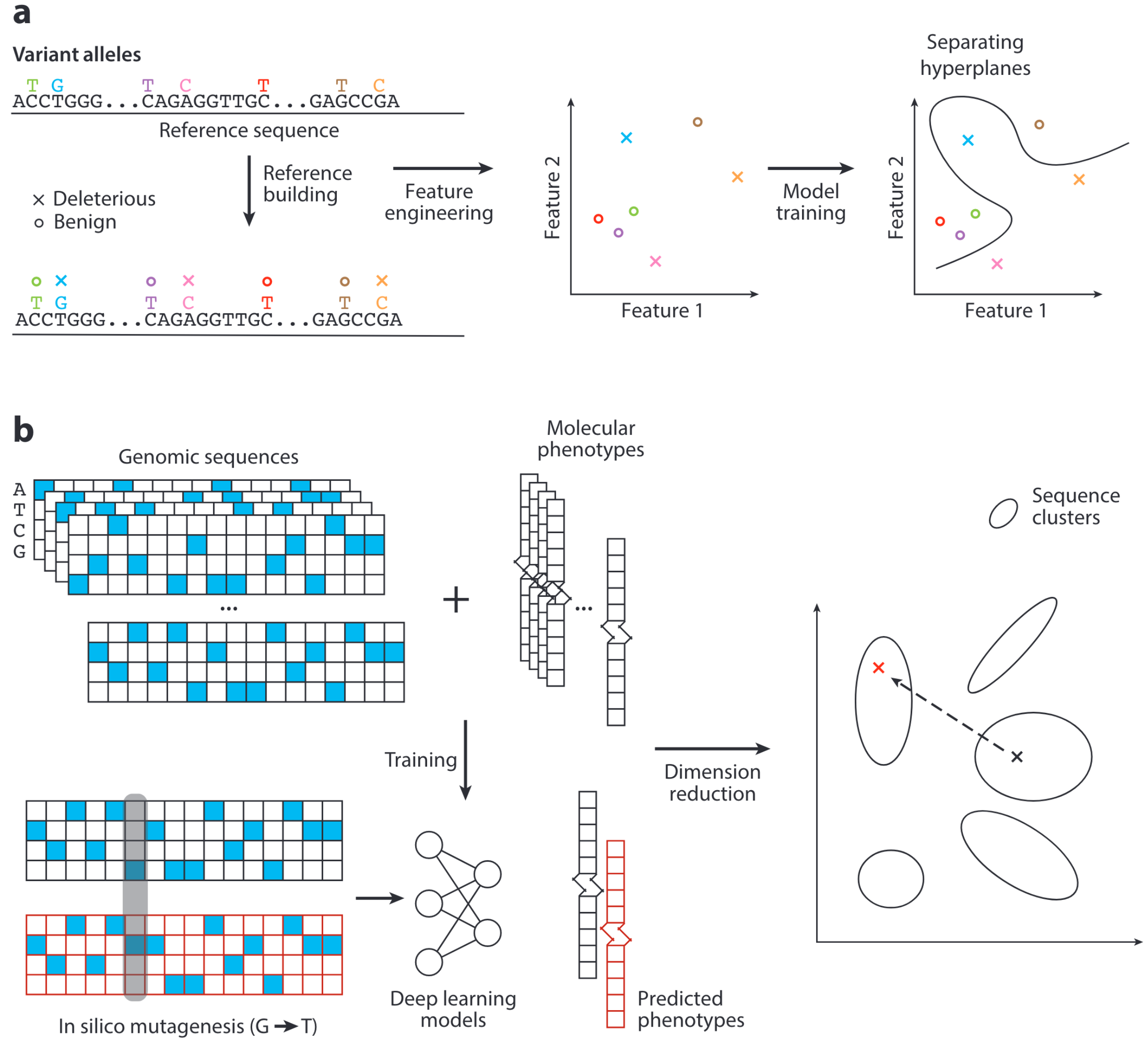
Finding Needles in the Haystack: Strategies for Uncovering Noncoding Regulatory Variants
Despite accumulating evidence implicating noncoding variants in human diseases, unraveling their functionality remains a significant challenge. Systematic annotations of the regulatory landscape and the growth of sequence variant data sets have fueled the development of tools and methods to identify causal noncoding variants and evaluate their regulatory effects. Here, we review the latest advances in the field and discuss potential future research avenues to gain a more in-depth understanding of noncoding regulatory variants.
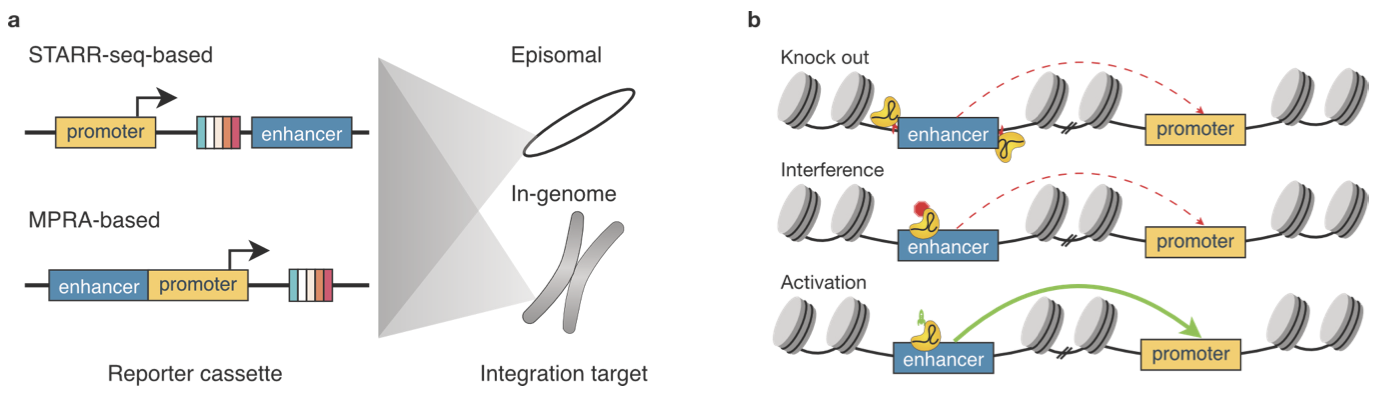
Functional genomic assays to annotate enhancer-promoter interactions genome wide
Enhancers are pivotal for regulating gene transcription that occurs at promoters. Identification of the interacting enhancer-promoter pairs and understanding the mechanisms behind how they interact and how enhancers modulate transcription can provide fundamental insight into gene regulatory networks. Recently, advances in high-throughput methods in three major areas-chromosome conformation capture assay, such as Hi-C to study basic chromatin architecture, ectopic reporter experiments such as self-transcribing active regulatory region sequencing (STARR-seq) to quantify promoter and enhancer activity, and endogenous perturbations such as clustered regularly interspaced short palindromic repeat interference (CRISPRi) to identify enhancer-promoter compatibility-have further our knowledge about transcription. In this review, we will discuss the major method developments and key findings from these assays.
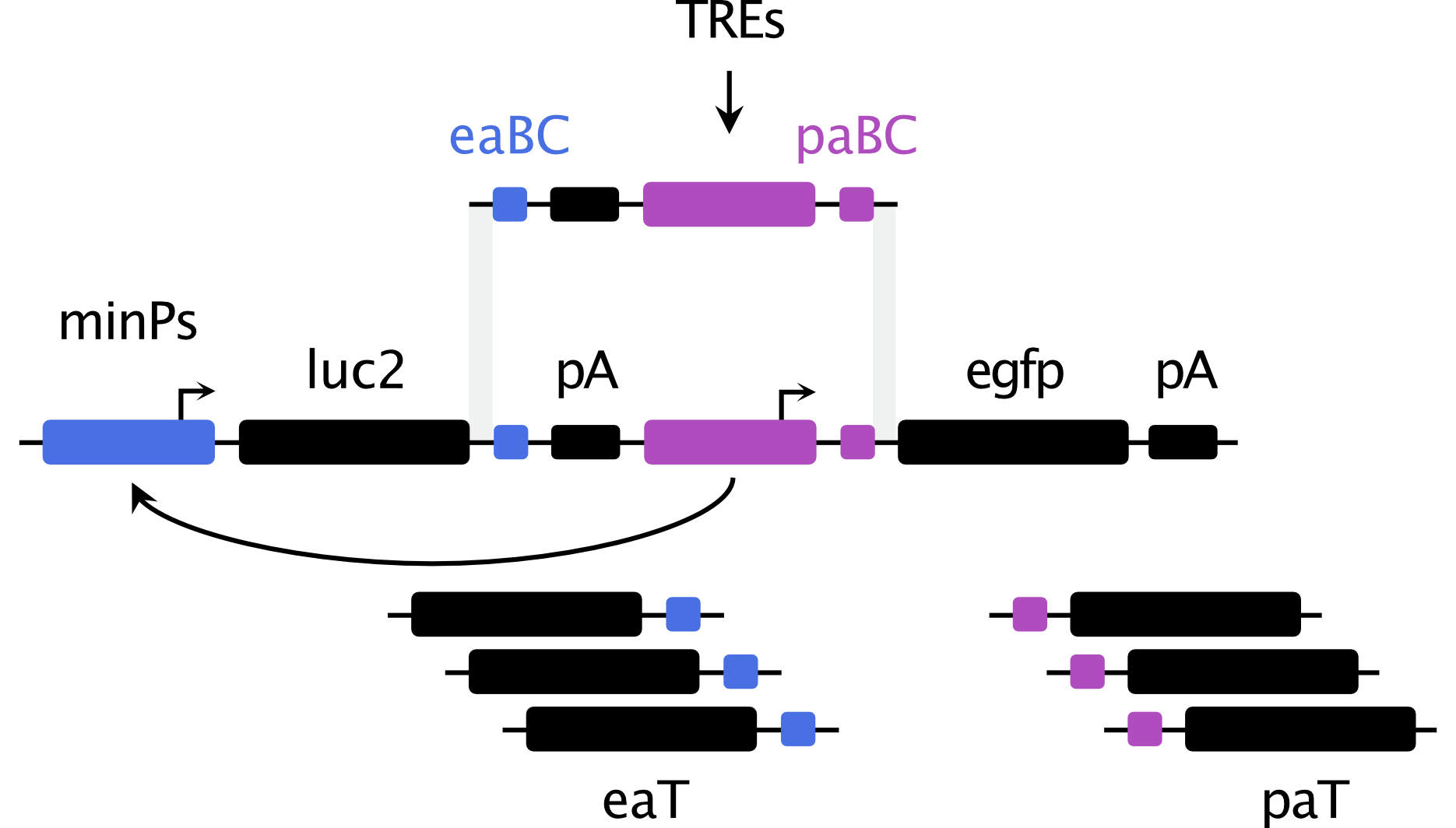
Dual promoter–enhancer activities reflect a unified regulatory logic
Growing evidence indicates that transcriptional regulatory elements can exert both promoter and enhancer activity; however, the relationship and determinants of this dual functionality remain poorly understood. We developed a massively parallel dual reporter assay that enables simultaneous assessment of the intrinsic promoter and enhancer potential exerted by the same sequence. Parallel quantification for thousands of elements reveals that canonical human promoters and enhancers can act as both promoters and enhancers under the same contexts, and that promoter activity may be necessary but not sufficient for enhancer function. We find that regulatory potential is intrinsic to element sequences, irrespective of downstream features typically associated with distinct element classes. Perturbations to element transcription factor binding motifs lead to disruptions in both activities, implicating a shared syntax for the two regulatory functions. Combinations of elements with different minimal promoters reveal reciprocal activity modulation between associated elements and a strong positive correlation between promoter and enhancer functions imply a bidirectional feedback loop used to maintain environments of high transcriptional activity. Finally, our results indicate that the magnitude and balance between promoter and enhancer functions are shaped by both intrinsic sequence properties and contextual regulatory influences, suggesting a degree of plasticity in regulatory action. Our approach provides a new lens for understanding fundamental principles of regulatory element biology.
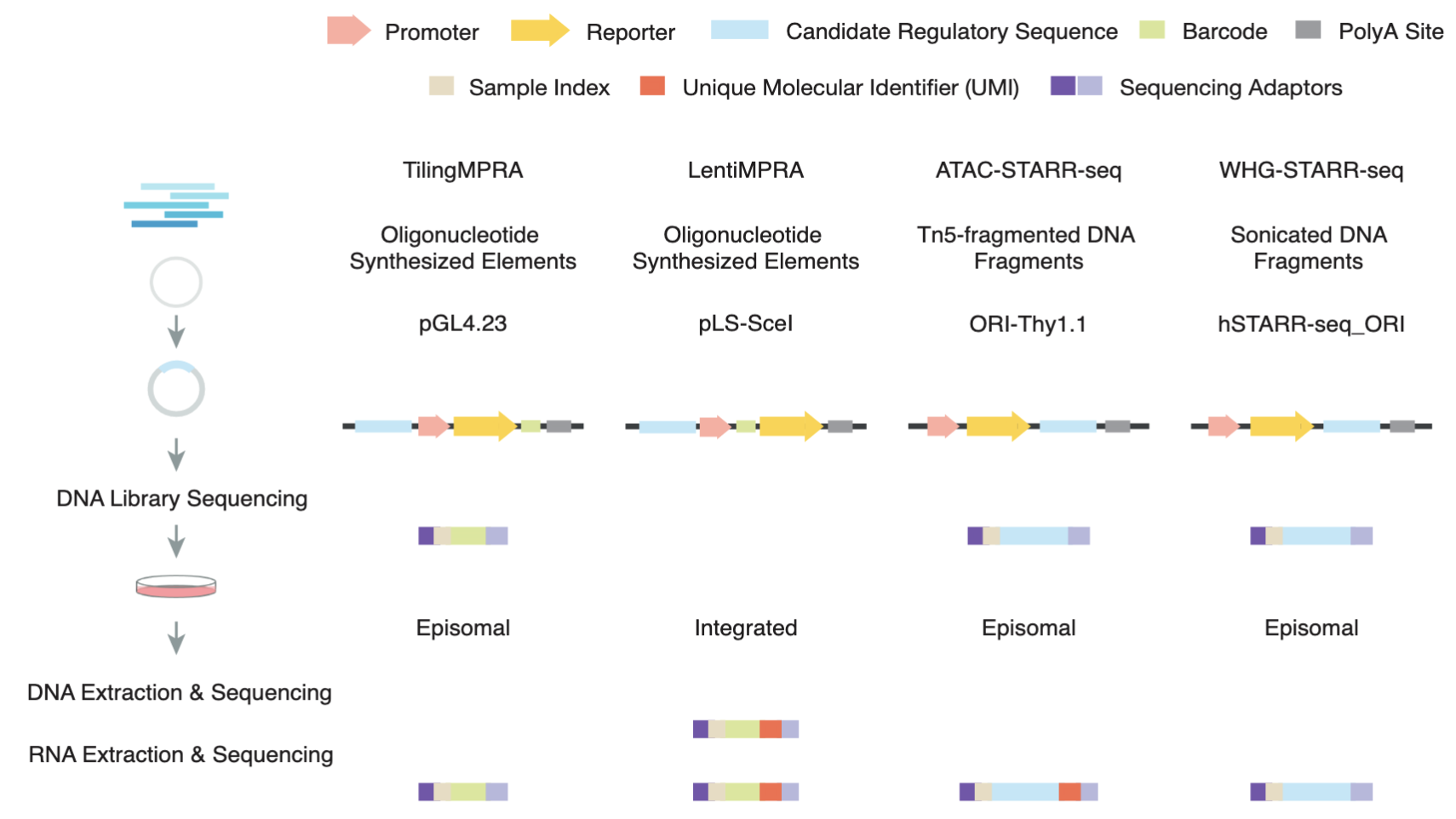
Comprehensive Evaluation of Diverse Massively Parallel Reporter Assays to Functionally Characterize Human Enhancers Genome-wide
Massively parallel reporter assays (MPRAs) and self-transcribing active regulatory region sequencing (STARR-seq) have revolutionized enhancer characterization by enabling high-throughput functional assessment of regulatory sequences. Here, we systematically evaluated six MPRA and STARR-seq datasets generated in the human K562 cell line and found substantial inconsistencies in enhancer calls from different labs that are primarily due to technical variations in data processing and experimental workflows. To address these variations, we implemented a uniform enhancer call pipeline, which significantly improved cross-assay agreement. While increasing sequence overlap thresholds enhanced concordance in STARR-seq assays, cross-assay consistency in LentiMPRA was strongly influenced by assay-specific factors. Notably, our results show that LentiMPRA exhibits a strong preference for promoter-associated sequences rather than enhancers. Functional validation using candidate cis-regulatory elements (cCREs) confirmed that epigenomic features such as chromatin accessibility and histone modifications are strong predictors of enhancer activity. Importantly, our study validated transcription as a critical hallmark of active enhancers, demonstrating that highly transcribed regions exhibit significantly higher active rates across assays. Furthermore, we show that transcription enhances the predictive power of epigenomic features, enabling more accurate and refined enhancer annotation. Our study provides a comprehensive framework for integrating different enhancer datasets and underscores the importance of accounting for assay-specific biases when interpreting enhancer activity. These findings refine enhancer identification using massively parallel reporter assays and improve the functional annotation of the human genome.
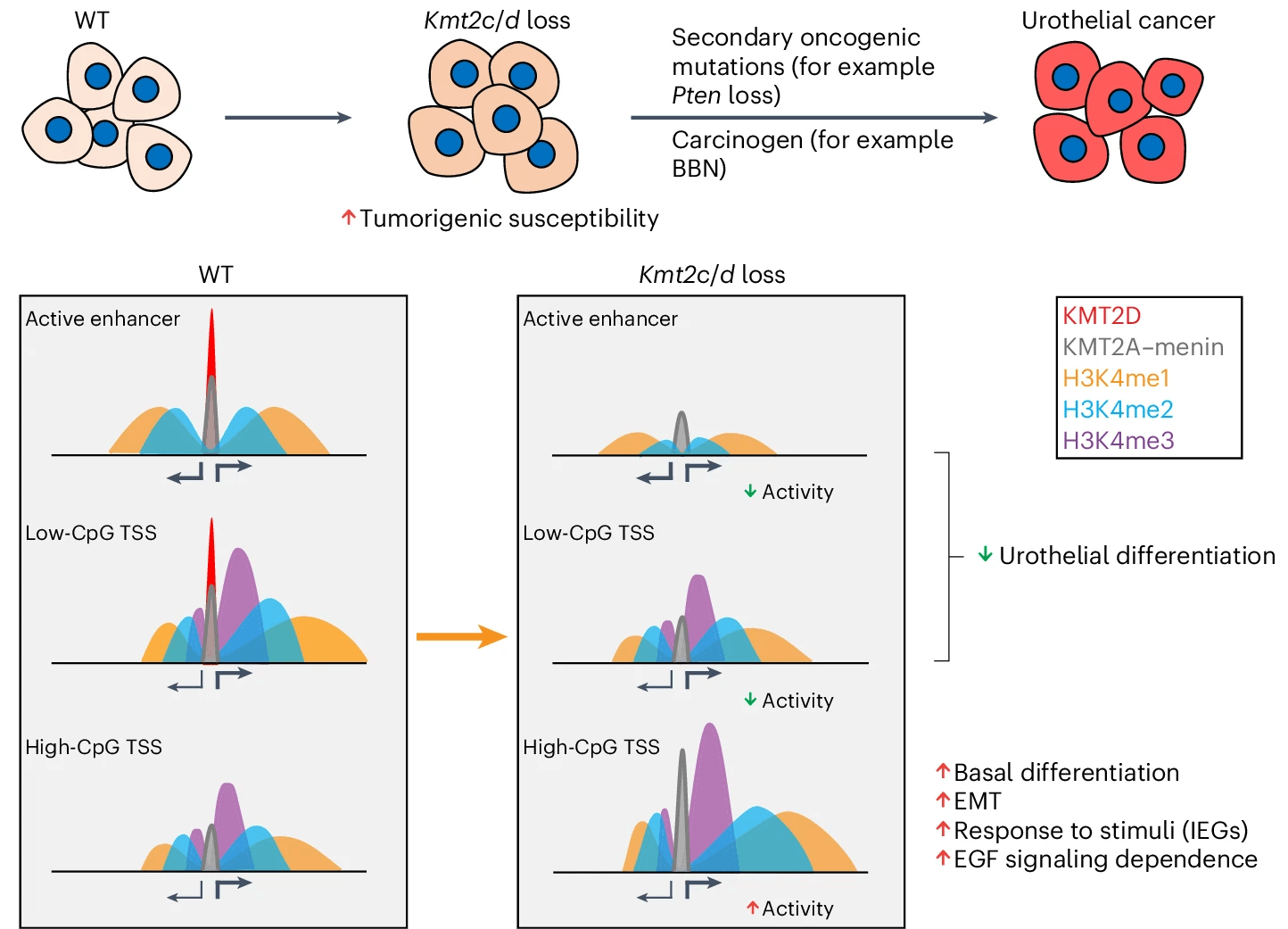
Loss of Kmt2c or Kmt2d primes urothelium for tumorigenesis and redistributes KMT2A–menin to bivalent promoters
Members of the KMT2C/D–KDM6A complex are recurrently mutated in urothelial carcinoma and in histologically normal urothelium. Here, using genetically engineered mouse models, we demonstrate that Kmt2c/d knockout in the urothelium led to impaired differentiation, augmented responses to growth and inflammatory stimuli and sensitization to oncogenic transformation by carcinogen and oncogenes. Mechanistically, KMT2D localized to active enhancers and CpG-poor promoters that preferentially regulate the urothelial lineage program and Kmt2c/d knockout led to diminished H3K4me1, H3K27ac and nascent RNA transcription at these sites, which leads to impaired differentiation. Kmt2c/d knockout further led to KMT2A–menin redistribution from KMT2D localized enhancers to CpG-high and bivalent promoters, resulting in derepression of signal-induced immediate early genes. Therapeutically, Kmt2c/d knockout upregulated epidermal growth factor receptor signaling and conferred vulnerability to epidermal growth factor receptor inhibitors. Together, our data posit that functional loss of Kmt2c/d licenses a molecular ‘field effect’ priming histologically normal urothelium for oncogenic transformation and presents therapeutic vulnerabilities.
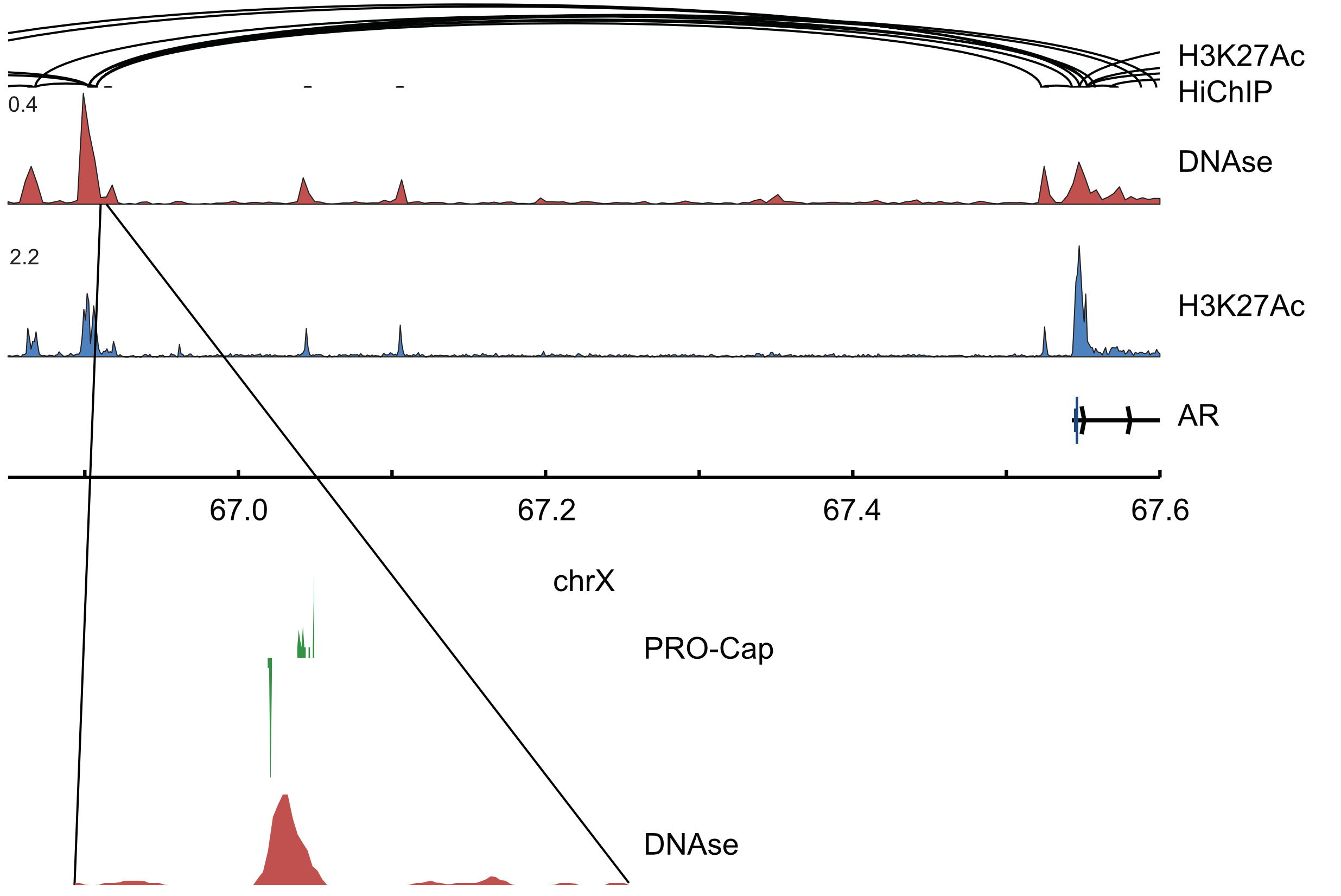
Capped nascent RNA sequencing reveals novel therapy-responsive enhancers in prostate cancer
AbstractMounting evidence suggests that enhancer RNA (eRNA) transcription start sites (TSSs) provide higher sensitivity and specificity for enhancer identification than histone modifications and chromatin accessibility. The extent to which changes in eRNA transcription correspond to changes in enhancer activity, however, remains unclear. Here, we used precision run-on and capped RNA sequencing (PRO-cap) to assess changes in enhancer activity in response to treatment with the androgen receptor signaling inhibitor, enzalutamide (ENZ). We identified 6,189 high-confidence candidate enhancers in the human prostate cancer cell line, LNCaP; 853 of which demonstrated significant changes in activity in response to drug treatment. Notably, we found that 67% and 54% of drug-responsive enhancers did not show similar changes in activity in previous studies that utilized ChIP-seq and ATAC-seq, respectively. Strikingly, 79% of regions with increased eRNA transcription showed no other biochemical alterations, implying that PRO-cap can capture a set of precise changes in enhancer activity that classical approaches lack the sensitivity to detect. We performed in vivo functional validations of candidate enhancers and found that CRISPRi targeting of PRO-cap-specific drug-responsive enhancers impaired ENZ regulation of downstream target genes, suggesting that changes in eRNA TSSs mark true biological changes in enhancer activity with high sensitivity. Our study highlights the utility of using PRO-cap as a complementary approach to canonical biochemical methods for detecting precise changes in enhancer activity and, in particular, for better understanding disease progression and responses to treatment.
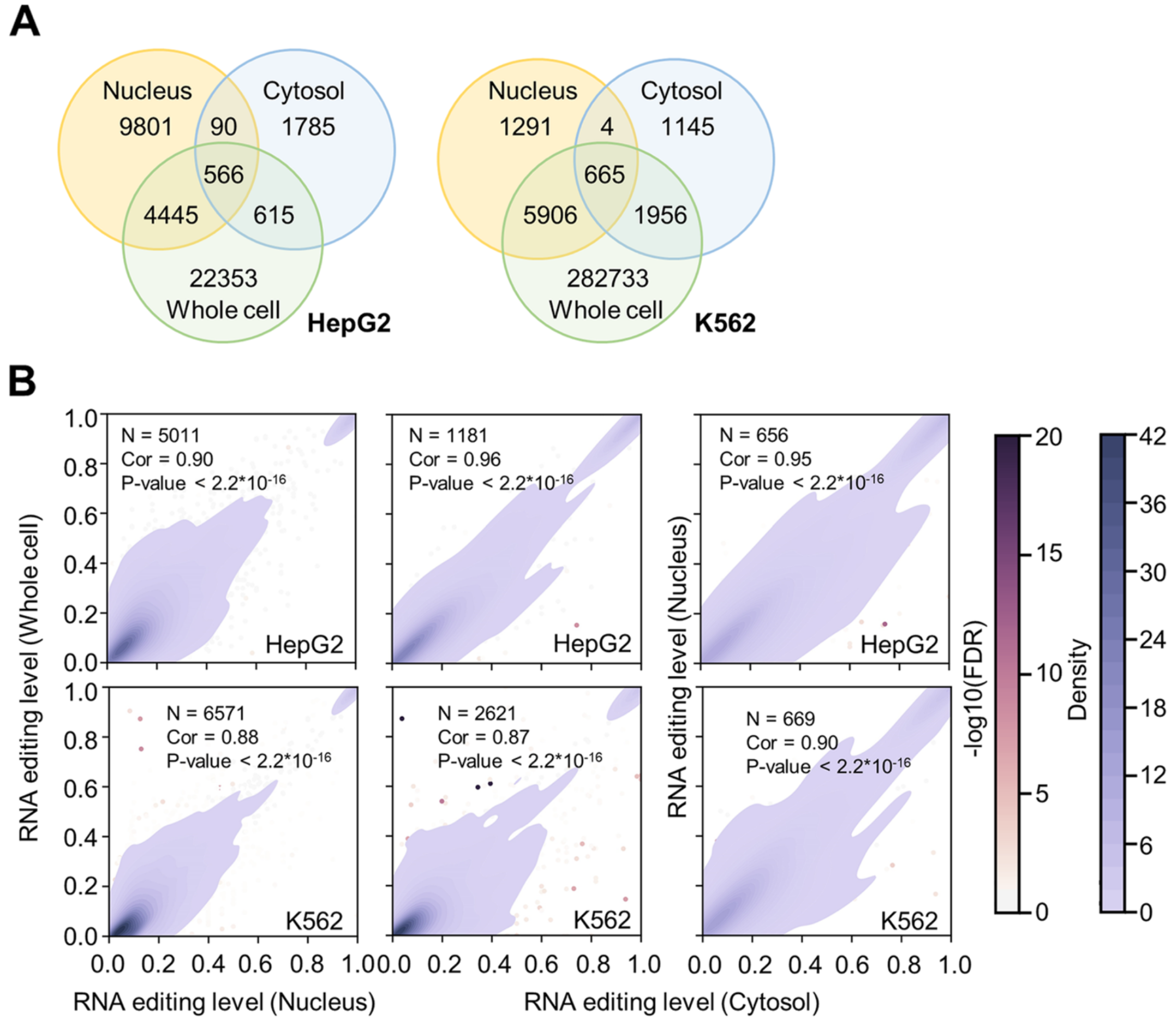
Survey of the binding preferences of RNA-binding proteins to RNA editing events
BACKGROUND: Adenosine-to-inosine (A-to-I) editing is an important RNA posttranscriptional process related to a multitude of cellular and molecular activities. However, systematic characterizations of whether and how the events of RNA editing are associated with the binding preferences of RNA sequences to RNA-binding proteins (RBPs) are still lacking.
RESULTS: With the RNA-seq and RBP eCLIP-seq datasets from the ENCODE project, we quantitatively survey the binding preferences of 150 RBPs to RNA editing events, followed by experimental validations. Such analyses of the RBP-associated RNA editing at nucleotide resolution and genome-wide scale shed light on the involvement of RBPs specifically in RNA editing-related processes, such as RNA splicing, RNA secondary structures, RNA decay, and other posttranscriptional processes.
CONCLUSIONS: These results highlight the relevance of RNA editing in the functions of many RBPs and therefore serve as a resource for further characterization of the functional associations between various RNA editing events and RBPs.
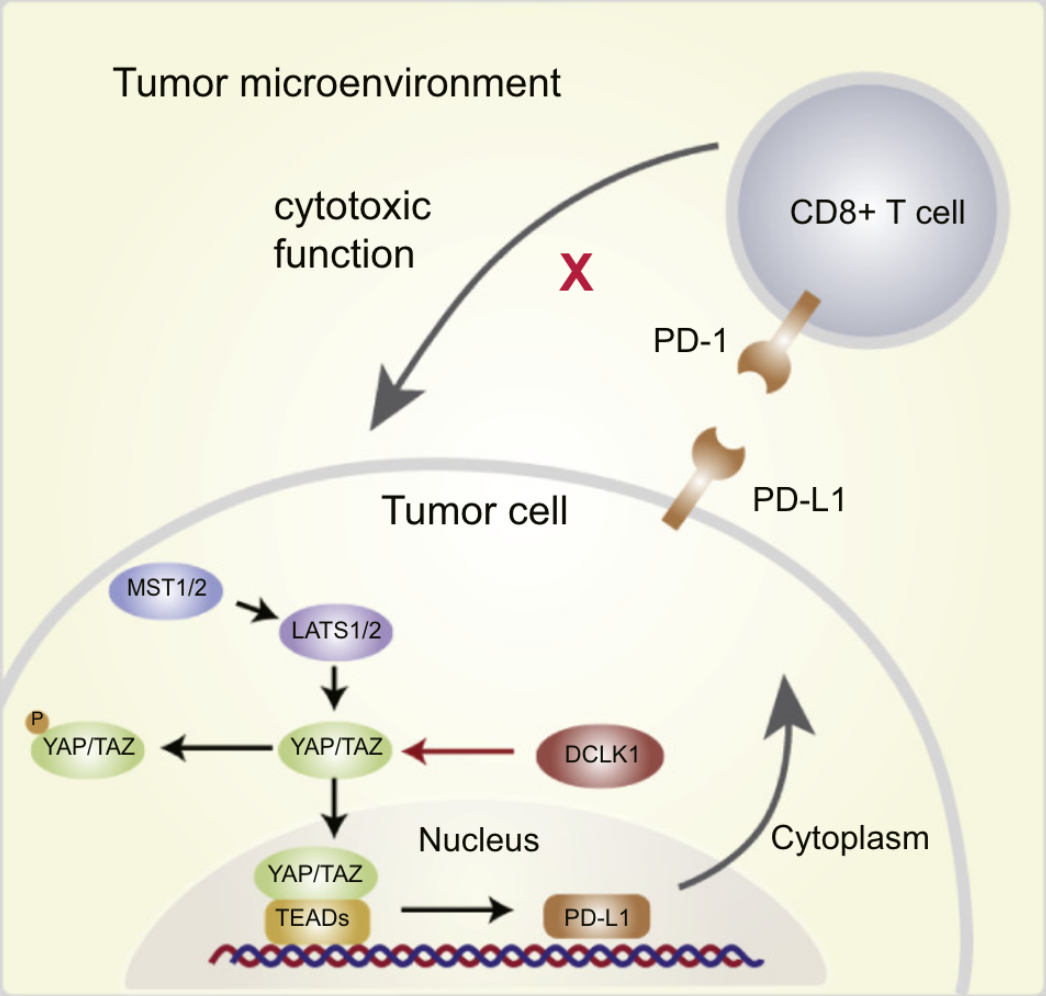
Inhibition of DCLK1 down-regulates PD-L1 expression through Hippo pathway in human pancreatic cancer
Immunotherapy is one of the most promising strategies for cancer, compared with traditional treatments. As one of the key emerging immunotherapies, anti-PD-1/PD-L1 treatment has brought survival benefits to many advanced cancer patients. However, in pancreatic cancer, immunotherapy-based approaches have not achieved a favorable clinical effect because of mismatch repair deficiencies. Therefore, the majority of pancreatic tumors are regarded as immune-quiescent tumors and non-responsive to single-checkpoint blockade therapies. Many preclinical and clinical studies suggest that it is still important to clarify the regulatory mechanism of the PD-1/PD-L1 pathway in pancreatic cancer. As a marker of cancer stem cells, DCLK1 has been found to play an important role in the occurrence and development of a plethora of human cancers. Recent researches have revealed that DCLK1 is closely related to EMT process of tumor cells, meanwhile, it could also be used as a biomarker in gastrointestinal tumors to predict the prognoses of patients. However, the role that DCLK1 plays in the immune regulation of tumor microenvironments remains unknown. Therefore, we sought to understand if DCLK1 could positively regulate the expression of PD-L1 in pancreatic cancer cells. Furthermore, we examined if DCLK1 highly correlated with the Hippo pathway through TCGA database analysis. We found that DCLK1 helped regulate the level of PD-L1 expression by affecting the corresponding expression level of yes-associated protein in the Hippo pathway. Collectively, our study identifies DCLK1 as an important regulator of PD-L1 expression in pancreatic tumor and highlights a central role of DCLK1 in the regulation of tumor immunity.
Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations
Each human genome carries tens of thousands of coding variants. The extent to which this variation is functional and the mechanisms by which they exert their influence remains largely unexplored. To address this gap, we leverage the ExAC database of 60,706 human exomes to investigate experimentally the impact of 2009 missense single nucleotide variants (SNVs) across 2185 protein-protein interactions, generating interaction profiles for 4797 SNV-interaction pairs, of which 421 SNVs segregate at > 1% allele frequency in human populations. We find that interaction-disruptive SNVs are prevalent at both rare and common allele frequencies. Furthermore, these results suggest that 10.5% of missense variants carried per individual are disruptive, a higher proportion than previously reported; this indicates that each individual's genetic makeup may be significantly more complex than expected. Finally, we demonstrate that candidate disease-associated mutations can be identified through shared interaction perturbations between variants of interest and known disease mutations.
bioSyntax: syntax highlighting for computational biology
BACKGROUND: Computational biology requires the reading and comprehension of biological data files. Plain-text formats such as SAM, VCF, GTF, PDB and FASTA, often contain critical information which is obfuscated by the data structure complexity.
RESULTS: bioSyntax ( https://biosyntax.org/ ) is a freely available suite of biological syntax highlighting packages for vim, gedit, Sublime, VSCode, and less. bioSyntax improves the legibility of low-level biological data in the bioinformatics workspace.
CONCLUSION: bioSyntax supports computational scientists in parsing and comprehending their data efficiently and thus can accelerate research output.
Circulating microRNAs: Promising Biomarkers Involved in Several Cancers and Other Diseases
Recently, many studies indicated that microRNAs (miRNAs) stably existed in various body fluids, including serum, plasma, saliva, and urine. Such miRNAs that exist in mammalian body fluids are known as circulating miRNAs, and they can transmit signals between cells and regulate intracellular gene expression. Currently, we barely understand the characteristics, sources, secretion, uptake, and functions of newly generated miRNAs. Particularly, it has been shown that certain types of circulating miRNAs can provide effective clinical data, suggesting their roles as novel biomarkers for the early detection of diseases such as cancers, cardiovascular diseases, and diabetes. Therefore, miRNAs have attracted much attention in academia for their promising applications in fundamental research and clinical diagnosis. This review summarizes some of the functional studies that have been conducted as well as the promising applications of circulating miRNAs, and we hope it will benefit other researchers in this field.